Fer un avió de paper és prou senzill; amb quatre plecs podem transformar un foli en un objecte volador. Però quantes formes diferents se t’acudeixen amb un sol paper? La papiroflèxia, també anomenada origami, és un art provinent del Japó que consisteix a realitzar plegaments a un full per donar-li forma tridimensional.
De la mateixa manera que donem forma als papers per recrear figures, les proteïnes necessiten estar plegades per dur a terme la seva funció.
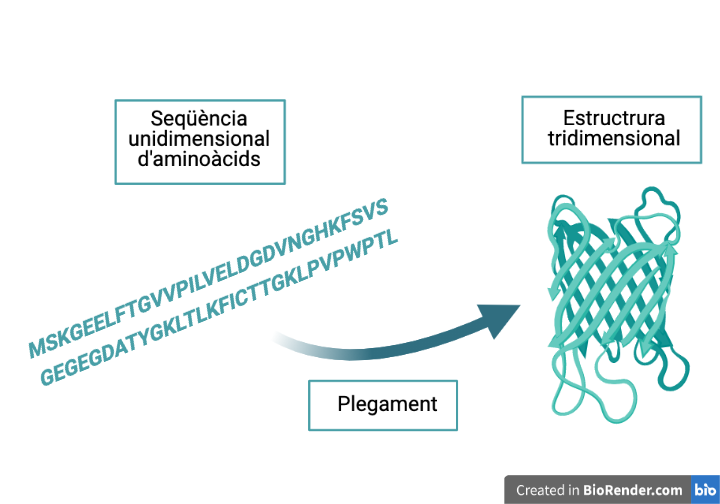
D’una cadena lineal a una estructura 3D
Les proteïnes són unes molècules que es troben en tots els organismes vius i que desenvolupen una gran varietat de funcions. Inclouen biomolècules tan famoses com l’albúmina de l’ou, la queratina de la pell o les hormones de creixement. Totes elles estan formades per una seqüència d’aminoàcids que podem representar amb la combinació de 20 lletres de l’alfabet.
Els aminoàcids tenen diferents propietats; per exemple, n’hi ha de més hidrofòbics i de més hidrofílics, o poden tenir diferent càrrega elèctrica, determinant així les interaccions que tenen entre ells. Recordem que per ser funcionals, les proteïnes han d’estar plegades. Però, com passem de la cadena lineal d’aminoàcids a l’estructura plegada tridimensional?
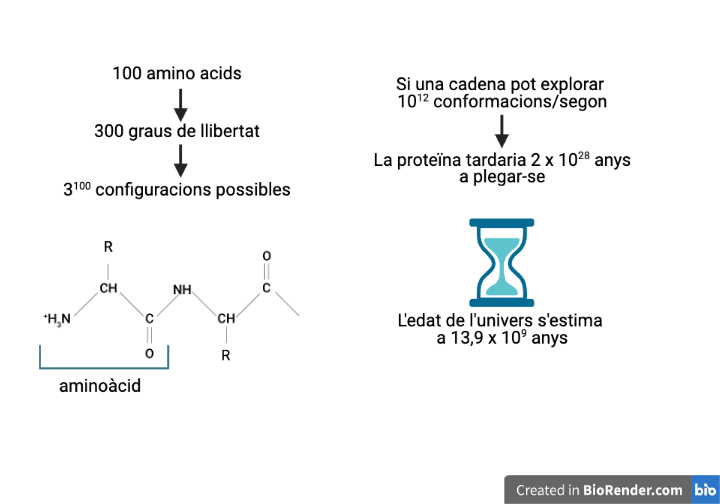
10000000000000000000000000000 anys són molts anys
Tal com va postular Christian Anfinsen a la recepció del Premi Nobel de Química l’any 1972, la cadena d’aminoàcids hauria de determinar l’estructura tridimensional de la proteïna. Tanmateix, si tenim en compte que cada aminoàcid presenta 3 graus de llibertat, corresponents als diferents angles de rotació, per combinatòria sabem que una cadena de 100 aminoàcids tindria 3¹⁰⁰ configuracions possibles. Suposant que la cadena pot explorar aproximadament 10¹² configuracions per segon, la proteïna tardaria 2×10²⁸ anys a plegar-se, que és més que l’edat de l’univers!
El mateix passaria si volguéssim determinar l’estructura tridimensional amb un ordinador per força bruta. Malgrat això, la proteïna aconsegueix trobar quin és l’estat energètic més favorable -l’estat natiu- entre totes les combinacions i plegar-se en segons o menys. Això és el que es coneix com a la paradoxa de Levinthal i és una de la raó per les quals és tan difícil trobar de manera computacional l’estructura tridimensional nativa de les proteïnes.
Determinar l’estructura tridimensional: un ventall d’aplicacions
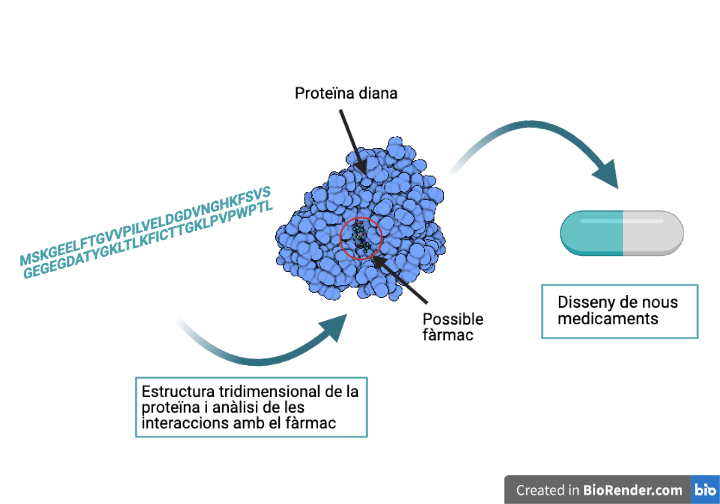
De manera experimental, podem conèixer l’estructura 3D de les proteïnes a partir de mètodes com la cristal·lografia de rajos X o l’espectroscòpia de ressonància magnètica nuclear (RMN). Paral·lelament, s’han realitzat diversos esforços per poder realitzar prediccions teòriques de l’estructura a partir de les forces físiques que existeixen entre les molècules. Tanmateix, encara no ens és possible predir-la acuradament a partir de la seqüència dels aminoàcids.
Conèixer l’estructura d’una proteïna a través de la seva seqüència tindria molts avantatges i aplicacions en diferents àmbits. En la biomedicina seria útil per poder estudiar les interaccions de les proteïnes diana amb els fàrmacs i agilitzar el disseny de medicaments i vacunes. Per altra banda, obriria noves possibilitats com ara dissenyar enzims per degradar residus industrials o molècules artificials per capturar carboni de l’atmosfera.
El concurs de papiroflèxia més difícil del món
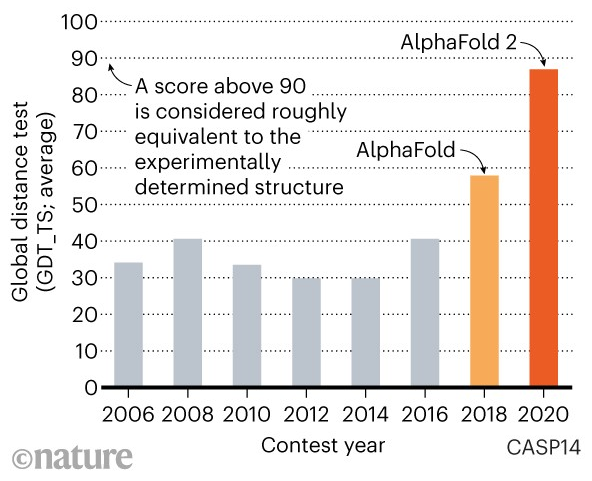
Desenvolupar mètodes per predir l’estructura d’una proteïna segueix sent un dels reptes més grans de la bioinformàtica, i és per això que biennalment es realitza l’experiment CASP. Aquest consisteix en un concurs científic en què s’envia la seqüència d’una proteïna amb una estructura desconeguda a centres de recerca. Els equips d’investigació han de realitzar prediccions de l’estructura tridimensional amb els mètodes computacionals que han desenvolupat. Fins fa quatre anys, la màxima precisió obtinguda en les prediccions rondava el 40%, però el passat novembre del 2020, l’empresa DeepMind va aconseguir una precisió superior al 85% utilitzant una intel·ligència artificial anomenada AlphaFold 2.
Què són les xarxes neuronals artificials?
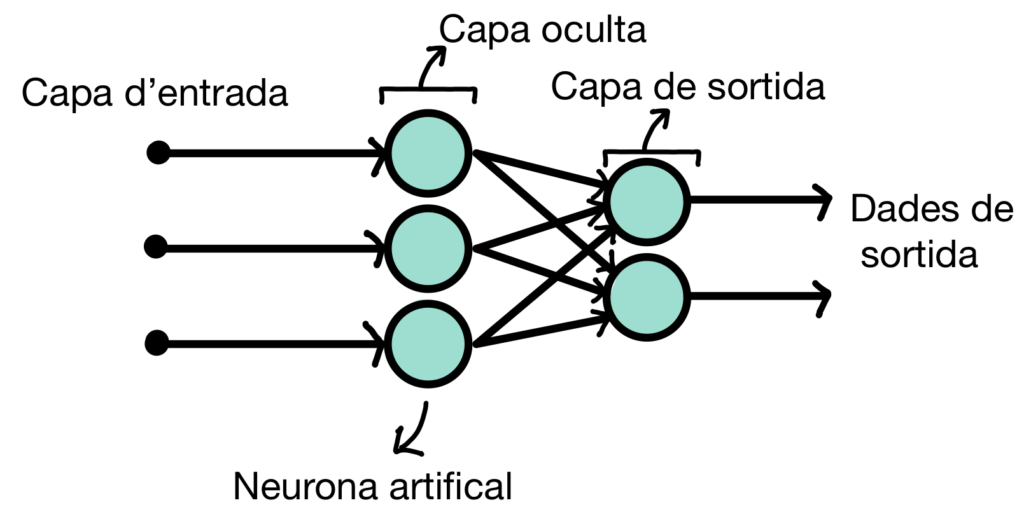
Els científics de DeepMind van utilitzar xarxes neuronals per poder realitzar prediccions de l’estructura tridimensional de les proteïnes a partir de la seva seqüència. Les xarxes neuronals artificials (XNA) són unes eines computacionals inspirades en les connexions del sistema nerviós dels animals. Permeten realitzar prediccions i tenen moltes aplicacions com ara el reconeixement de veu, fer prediccions del temps atmosfèric o estudiar el mercat financer.
Les xarxes neuronals artificials estan formades per un conjunt d’unitats que estan interconnectades entre si i que anomenem neurones artificials. Alhora, aquestes neurones s’organitzen en unes capes ocultes tot formant una xarxa. A partir d’una base de dades, la xarxa pot ser entrenada per predir un resultat variant els pesos de les connexions entre les neurones artificials. El sistema estructurat de neurones permet que la xarxa aprengui i es reorganitzi a mesura que li donem dades per entrenar-se.
La intel·ligència artificial al rescat
Les xarxes neuronals es poden perfeccionar i s’hi poden afegir nous elements que permeten realitzar prediccions més complexes. En aquest cas, DeepMind ha desenvolupat el programa Alpha Fold 2, que intenta resoldre el dificilíssim problema de determinar l’estructura 3D de proteïnes a partir únicament de la seva seqüència aminoacídica. El programa s’ha entrenat amb més de 170.000 estructures de la base de dades Protein Data Bank i presenta resultats optimistes. A més, Alpha Fold 2 ja s’ha utilitzat per realitzar prediccions de l’estructura de molècules desconegudes com ara sis proteïnes poc estudiades del virus SARS-CoV-2.
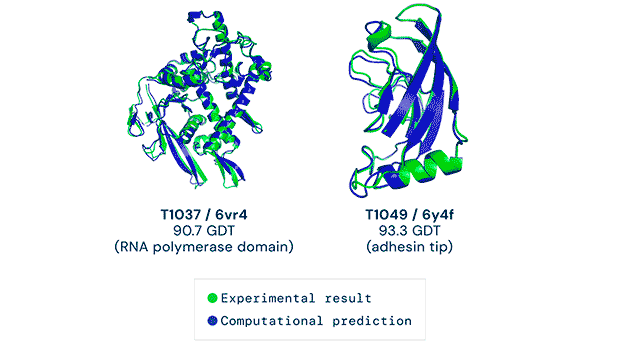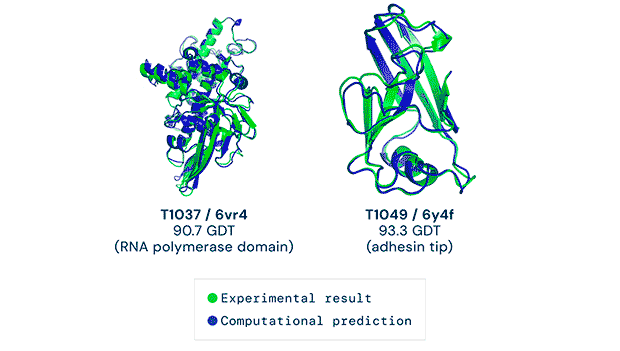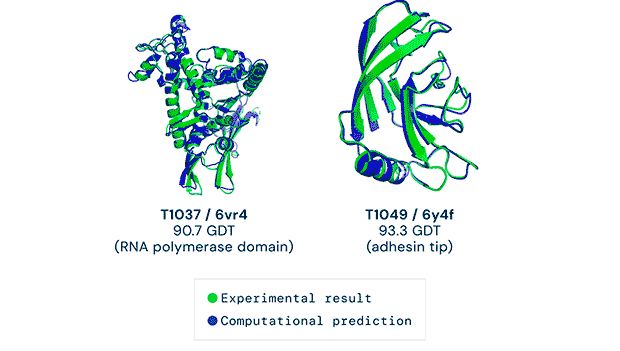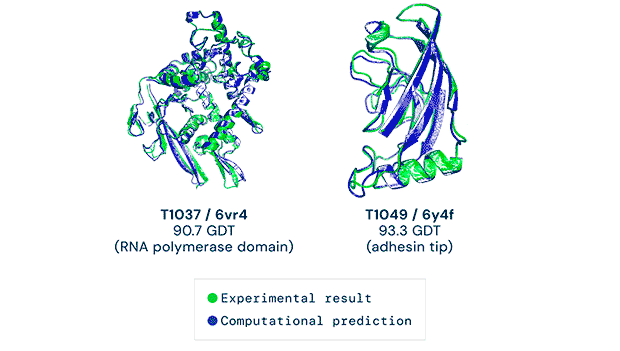
Ja per acabar…
Els avenços en l’adquisició de dades en els camps de la biologia i la biomedicina ens permeten, avui en dia, recollir una gran quantitat d’informació que ni tan sols tenim temps d’analitzar de manera tradicional. La intel·ligència artificial, però, està esdevenint una eina molt poderosa, ja que ens permet reconèixer patrons, fer prediccions i analitzar el gran volum de dades generat.
Els resultats obtinguts per DeepMind en el que podríem considerar el joc de papiroflèxia més difícil del món marquen un abans i un després en la biologia estructural, obrint camí a noves investigacions que tindran un gran impacte en la ciència i en la medicina.
En un futur no molt llunyà, la intel·ligència artificial ens permetrà solucionar problemes tan difícils i importants com realitzar diagnòstics mèdics més acurats, predir la funció de proteïnes desconegudes o analitzar imatges biomèdiques de manera automàtica.
Per saber-ne més
DeepMind – High accuracy protein structure prediction using Deep Learning
Nature – “‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures.”
Journal de Chimie Physique – Are there pathways for protein folding?